
Lawmakers and patients have called for scrutiny of the Food and Drug Administration’s medical device recall oversight after several high-profile recalls in the last five years. The U.S. Government Accountability Office said in January it would look into the agency’s oversight of medical device recalls, a little over a decade after a previous investigation by the watchdog found the FDA often failed to conduct recall-related inspections and document why a recall was terminated.
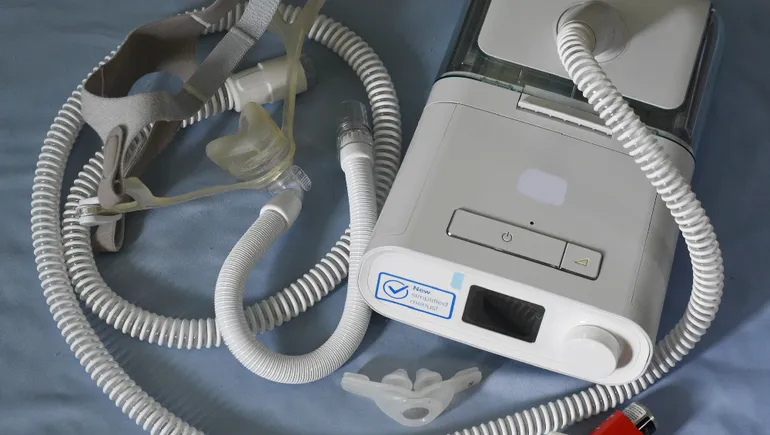
Recent safety problems with devices have gotten lawmakers’ attention again. Philips is still resolving a recall of more than 15 million sleep apnea machines and ventilators due to health risks from soundproofing foam breaking down, and the company recently halted sales of the devices in the U.S. Problems with inaccurate readings from pulse oximeters have also garnered scrutiny, with some agency advisors calling for faulty products to be recalled.
MedTech Dive interviewed product and patient safety experts about what changes the FDA can make to improve the recall process. Here are six recommendations:
1. Improve tracking of adverse events
Adverse event reports are important for informing the FDA’s oversight of recalls. Manufacturers are required to submit reports of serious injuries, deaths or device malfunctions that could cause a health risk to the FDA. Hospitals, ambulatory surgical centers and nursing homes must also submit reports of suspected medical-device-related deaths.
This process has two problems: manufacturers may delay sending reports to the FDA, and physician offices, where certain procedures may be done, are not required to report problems with devices, said Madris Kinard, a former FDA analyst who built software called Device Events to track adverse events and recalls.
Before Philips’ current recall of respiratory devices, an FDA inspection found the company held thousands of reports of problems with its sleep apnea machines and ventilators. Manufacturers are required to submit these reports within 30 days, or as quickly as five days when the risk of harm to the public is “substantial,” but Philips was years late in submitting several reports, according to a Propublica article.
“They found so many complaints that hadn’t been submitted to the FDA, and then of course, they just flood in after that inspection,” Kinard said. “With [continuous positive airway pressure machines], it got a lot of attention, so the reports came in. We’ll see that with other devices, and then we’re waiting and don’t see the reports come in.”
Bayer’s Essure device, which was pulled from the U.S. market in 2019, also had thousands of complaints that were not reported to the FDA in a timely fashion, according to the International Consortium of Investigative Journalists.
One solution could be adding a liaison position where if a site doesn’t meet quality standards in an inspection, someone is assigned to go between the inspection and the FDA to make sure things get resolved, Kinard said.
“Otherwise, they just wait, go out for another inspection in two, three years and see the same things again,” she added.
Kinard also noted that even if a problem is thought to be caused by user error, it still needs to be reported to the FDA. Maybe the instructions need to be updated or there is a problem with the device. For example, after the FDA flagged a growing number of adverse events related to surgical staplers in 2019, Medtronic recalled millions of staplers that were missing a pin.
Physician offices should also be counted under the user facilities that are required to submit medical device reports to the FDA, Kinard said. Breast implants are often removed in a physician’s office instead of a hospital, so reports of problems with the devices might not be sent to regulators. Even if physicians are reporting information to an implant registry, most registries don’t report regularly to the FDA, Kinard said.
FDA spokesperson Kristina Wieghmink wrote in an email that medical device reports are used to monitor device performance, detect potential safety issues and contribute to the benefit-risk assessment of products.
She confirmed that while the FDA generally requires manufacturers to submit reports within 30 days, there are instances where the agency may grant an exemption or variance.
“The FDA will investigate potential violations of the applicable laws and take action as appropriate,” Wieghmink wrote.
2. Require electronic recall notifications
At the start of a recall, manufacturers often mail out notifications to hospitals in overnight envelopes, even though the FDA has issued guidance encouraging the use of electronic communications. Relying on paper notices alone can slow the process down as mail is routed through hospitals and as manufacturers await a mailed acknowledgment back, said Guillermo Ramas, CEO of Notisphere, a company that makes software to manage medical recalls.
It can take about two weeks between when a recall is issued and when a recall coordinator at a hospital knows about it via paper, Ramas said. Hospitals then have to manually input the recall information into their internal databases. The process can repeat itself if a device company doesn’t receive enough acknowledgments back from hospitals.
“You’re now six months into a recall where there’s very little visibility into is everybody aware of the recall? Is every hospital pulling items from the shelves and stopping the use of them?” Ramas said.
Despite the FDA’s guidance, many manufacturers still use paper communications.
“There’s a little bit of a disconnect there,” Ramas said. “I’ve had suppliers straight up tell me, ‘No, the FDA doesn’t allow it electronic; it needs to be paper.’ It couldn’t be further from the truth.”
Ramas sees a missed opportunity where the FDA could take a more active role in educating manufacturers and encouraging them to use a more efficient process.
“If you’re a recall coordinator at a multi-state health system, think about the task you have in front of you now. You have 23 pages of paper with serial numbers, and you’re supposed to be telling people in hospitals over five states to go check.”

Guillermo Ramas
Notisphere CEO
Another challenge is that critical information in these notices, such as what lots were affected or the distribution dates for the recalled devices, are sent out as unstructured data.
“Imagine … a hospital receiving a paper alert that shows 23 pages of serial numbers,” Ramas said. “This is happening every day. So, if you’re a recall coordinator at a multi-state health system, think about the task you have in front of you now. You have 23 pages of paper with serial numbers, and you’re supposed to be telling people in hospitals over five states to go check.”
A hospital supply chain industry group issued a report in 2021 that found healthcare providers spent from 16 to 104 hours per recall. One provider said they processed 42 Class I recalls in a year, costing more than $20,000, according to the report from the Association for Health Care Resource & Materials Management (AHRMM).
In the worst-case scenario, physicians don’t receive recall information at all or the affected devices aren’t isolated.
“We will see devices get implanted after a recall happens,” Kinard said.
Sen. Richard Durbin, D-Ill., who requested the GAO’s review, introduced a bill last year that would require manufacturers to send out electronic recall notifications and require the FDA to maintain an electronic, public database of recall notifications.
“I fail to understand why that can’t come straight from the FDA,” Ramas said.
3. Improve the use of UDIs
Unique device identifiers, labels with specific codes assigned to a medical device, can make products easier to track in the event of a recall. The FDA published a final rule to establish a UDI system in 2013, and manufacturers now incorporate UDIs in the label, as required. But hospitals and payers have been slower to adopt the system, making it “functionally useless,” said Kushal Kadakia, a student at Harvard Medical School and first author of a paper in Health Affairs calling for more accountability of the device recall process.
“We have made this problem unique to devices, because we do track medications. We have national drug codes. We have done it for decades,” Kadakia said. “There’s no reason, to me, that a product that is just as important to my clinical practice and to patient care is for some reason erased.”

A sample UDI by the FDA shows information included in the label.
UDI information is still not included in many recall notices, and the data is often in an unstructured format. Just a quarter of Class I recall notices between 2018 and 2022 contained UDI information, according to the Health Affairs paper.
In a recall, that means hospital staff must manually check if a recalled lot is on the shelf instead of locating the affected devices in an electronic inventory. To make UDIs work better for hospitals, AHRMM recommends ensuring UDIs are tracked in the recall notice, as well as a health system’s electronic health record and enterprise resource planning software. Providers also said including UDI information would be of “minimal benefit” if it was communicated in a text-based format such as a paper or PDF notice.
For patients, UDIs are critical to know if a device or implant they received has been recalled. For example, when Allergan recalled some of its textured breast implants due to the risk of a type of lymphoma, many people weren’t even aware they had a recalled implant until they were replacing or removing it, said Maria Gmitro, president and founder of the Breast Implant Safety Alliance.
“Adoption of something like a UDI is so important because these devices went by different names,” Gmitro said. “As a patient, even if I saw FDA recalled Allergan Biocell, well I didn’t have Allergan, I had McGhan. They don’t realize that the names actually changed.”
Doctors may not be the ones who communicate about a recall either, as they might not be aware of a recall, or it gets lost in the shuffle between patient address changes, Gmitro said.
One solution could be to track UDIs on claims forms, adding an incentive for them to be tracked in practice, Kadakia said. Standards group X12 added UDIs to Medicare claims forms in a 2022 update, but the National Committee on Vital and Health Statistics, which advises the Department of Health and Human Services, rejected several updates to the forms.
4. Faster and more transparent recalls
After a manufacturer starts a recall, it can take weeks or months for the FDA to post a notice. One reason for the delay is that the FDA is working to classify a recall to determine how serious or widespread the problem is, denoting Class I for the highest-risk events.
“When it takes six to eight weeks, three months to get a Class I recall issued, even through the voluntary process, then clearly something isn’t working well,” said Teresa Murray, who leads the consumer watchdog office for public interest group PIRG. “There needs to be a way for that to happen as soon as possible because, again, people’s lives are at stake.”
The FDA updates its medical device recall database after it classifies a recall and later after it terminates a recall, Wieghmink said. Companies are required to submit a written report to the agency when they initiate a correction or removal of a device. The FDA reviews the company’s strategy to address the problem, assesses the health hazard presented by the product and determines if the problem violates statutes or regulations before classifying the recall.
“When it takes six to eight weeks, three months to get a Class I recall issued, even through the voluntary process, then clearly something isn’t working well.”
Teresa Murray
Consumer Watchdog for PIRG
The language in the notices themselves is geared toward providers and can be very technical, said Shannon Davila, executive director of total systems safety at the safety group ECRI.
“There’s a lot of ambiguity sometimes when these recall notices go out to patients and providers,” Davila said. “It’s unclear what the next steps are, who they’re supposed to follow up with and contact.”
Davila recommended that at-home devices have clear instructions for how patients can register them so they can get notified in the event of a recall, and that providers also communicate the importance of registration.
The duration of recalls is also growing. The GAO’s inspection in 2011 found the time between when a recall started and was terminated was an average of about 17 months for Class I recalls.
A group of researchers recently took a closer look at Class I recalls posted between 2018 and 2022. They found that it took a median of 24 months from recall initiation to termination, and 10 recalls had been going on for more than three years.
“One thing that has really struck our team is that the time it takes to resolve a recall — from when GAO mapped it in the report in 2011 compared to when our team analyzed it last year — has increased significantly,” Kadakia said.
The FDA’s Wieghmink said the agency closely monitors recalls that remain open and requests companies to submit periodic status reports.
It’s difficult to know what contributed to the longer times because data about the steps of the process aren’t publicly available. The reason why a recall was considered terminated by the FDA also isn’t public — a fact highlighted by the GAO report.
“It also begs the question of, should we have clearer metrics that are publicly accountable?” Kadakia said.
5. Strengthen premarket reviews
The majority of medical devices go through the FDA’s 510(k) pathway, where manufacturers rarely need to do premarket clinical testing, as they merely need to prove that a device is substantially equivalent to a product already on the market, called a predicate device. The authors of the Health Affairs paper noted that most Class I recalls affect devices that have been authorized through the 510(k) pathway.
“The best way to avoid recalls is to have better premarketing standards,” said Michael Abrams, a senior health researcher with consumer advocacy group Public Citizen.
He described the situation as a “house of cards,” with predicates allowing cleared devices to be based on technology that is decades old. Meaning, recalls could have wider consequences, because if a company is recalling one device based on a predicate, it could potentially implicate others that were also based on that same product.
Abrams cited reports of pulse oximeters giving misleadingly high blood oxygen readings to patients with darker skin as an example of the problem. Most pulse oximeters were cleared through the FDA 510(k) pathway, he said. An advisory panel recently recommended new pulse oximeters be required to provide clinical data, with trials that include more participants across all skin tones.
“We think the FDA should be getting more aggressive about looking at which of these devices are failing in that way and recalling them,” Abrams said.
Draft guidance published by the agency in September 2023 could help address part of the problem. In the documents, the FDA advised companies to choose predicate devices that have been cleared using well-established methods, met safety and performance standards and don’t have design- or use-related safety problems. The agency also described scenarios when clinical data may be needed to demonstrate substantial equivalence to a predicate.
6. The FDA should use its recall authority
The FDA has the authority to mandate medical device recalls, but the vast majority are voluntary.
“They have the ability to enforce and to push and yet they don’t seem to do it that way,” Notisphere’s Ramas said, adding that the agency might take more of a neutral ground because it wants to be seen as collaborative with manufacturers and health systems.
Kinard, of Device Events, said one reason most recalls are called voluntary is to protect the agency from liability.
“When you’re communicating about a recall, it has to be strong or else people don’t act,” she said. “If they think it’s voluntary, the manufacturers use that to say, ‘We’re just doing this out of an abundance of caution.’”
Wieghmink said that most recalls are considered voluntary, but the FDA has the authority to take action if a manufacturer fails to do so, particularly if there’s reasonable probability it would cause serious adverse health consequences or death.
“However, in practice, the FDA has rarely needed to require a medical device recall,” Wieghmink wrote. She added, “Voluntary recalls remain the quickest, most effective way to remove or correct a medical device, its labeling, or marketing in the U.S. until issues are fully resolved.”
“When you’re communicating about a recall, it has to be strong or else people don’t act.”
Madris Kinard
Device Events CEO
Abrams and Kadakia called for the FDA to get more resources for handling recalls quickly. Kadakia proposed including recall performance goals in the next medical device user fee agreements, which set how much the agency can collect from manufacturers in fees.
Abrams called for more direct funding from Congress rather than fees collected from the industry. When problems arise, the FDA should be able to work with manufacturers and providers to find the faulty devices, and recall or remove them from the market, he said.
“That does require resources,” Abrams said. “And it requires that the FDA have the authority to do it without fear of retribution from the manufacturers that because of user fees, pay a substantial portion of their operating budget.”