
Dive Brief:
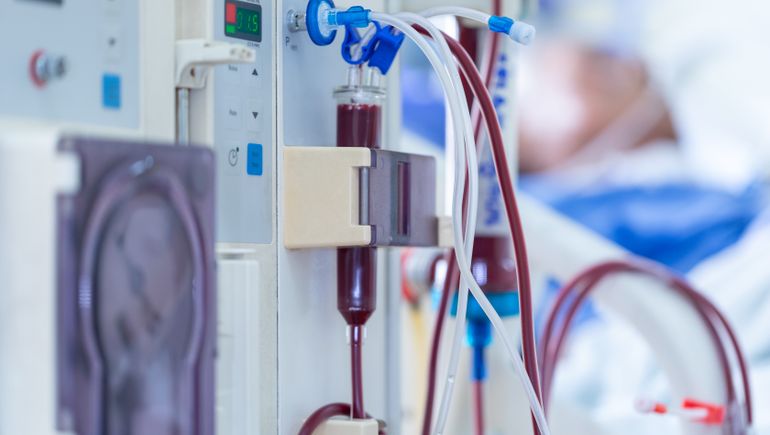
- Medtronic’s recall of hemodialysis catheters was categorized by the U.S. Food and Drug Administration as a Class I event, according to a Friday entry in the FDA’s database.
- The company contacted customers in June after routine manufacturing testing identified a blockage that could obstruct the catheter, potentially delaying treatment and leading to outcomes such as blood clots and the destruction of red blood cells.
- Healthcare providers with devices covered by the recall should immediately quarantine and stop use of the catheters. Medtronic has received no confirmed complaints related to the problem, and no reports of adverse events or deaths.
Dive Insight:
The Medtronic recall affects specific lots of Mahurkar Acute Triple Lumen Catheters and Mahurkar Acute High Pressure Triple Lumen Catheters. Medtronic’s Mahurkar Elite Catheters are unaffected by the recall.
“Medtronic initiated a voluntary recall related to Mahurkar Acute Triple Lumen Catheters and Mahurkar Acute High Pressure Triple Lumen Catheters because a blockage in the catheter was identified during routine manufacturing testing which could lead to delay of treatment and/or reduced flow or particulate dislodgement that may result in hemolysis, embolism/embolus, or thrombosis/thrombus,” a company spokesperson wrote in an email.
The urgent field safety notice Medtronic sent in June provides further details. Medtronic’s investigation revealed that excessive MDX, a silicone-based lubricant which coats the catheter tip, can block the central catheter lumen. Some of the risks relate to the potential for uncured or excessive MDX to dislodge.
Medtronic, which is working with IQVIA’s Novasyte on the recall, previously reported a potential hub defect with its Mahurkar Acute Dual Lumen High Flow (13.5 French) Hemodialysis Catheters that could cause arterial and venous blood to mix. That problem prompted the FDA to publish a Class I recall notice in January.