
A decade of progress has brought hope to management of genetic tumor-causing conditions that fall under the umbrella of neurofibromatosis (NF).
“There have been more discoveries and more approaches in the past decade than the 100 years before that,” said Roger J. Packer, MD, of the Gilbert Family Neurofibromatosis Institute at Children’s National Hospital in Washington, D.C. “So it’s really been very exciting.”
Before, “other than diagnosing these children, I couldn’t offer them very much,” Packer explained in an interview where a media relations person was present. Now though, “we’ve been able to offer them life-altering therapies.”
NF is a fairly diverse group of conditions in which mutations lead to widespread growth of tumors on nerves. Though still considered a rare disease, altogether they affect roughly one in 2,000 persons, noted Annette Bakker, PhD, president of the Children’s Tumor Foundation, a nonprofit focused on NF research and education.
Type 1
The most common type is NF1, which affects about one in every 3,500 children born. It is caused by a mutation in the NF1 gene on chromosome 17 that causes loss of neurofibromin and activation of a signaling pathway that results in cellular overgrowth, leading to both benign and high-grade cancers. Clinical manifestation at birth or early childhood can result in blindness, deafness, bone abnormalities, disfigurement, learning disabilities, disabling pain, and cancer.
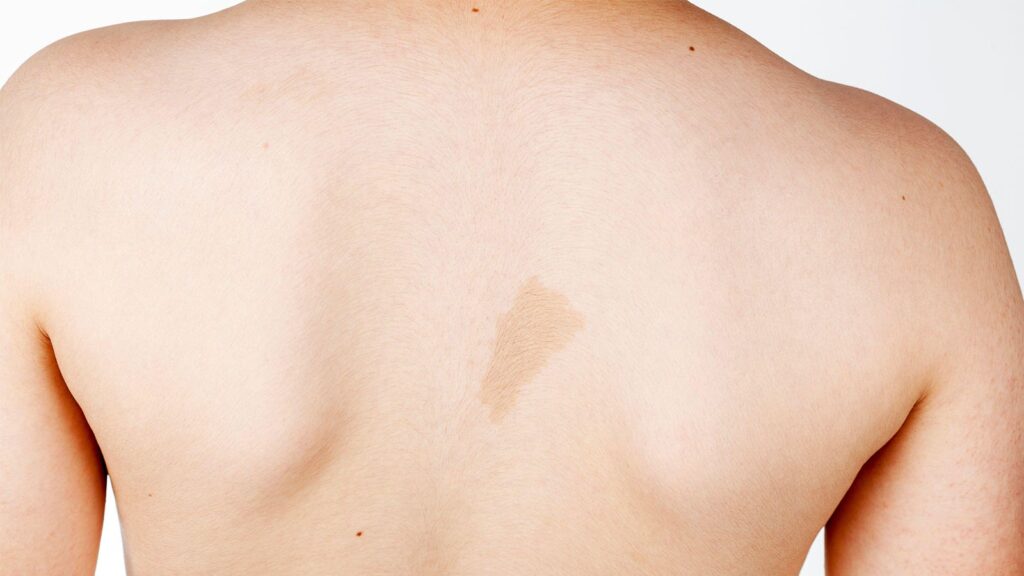
Diagnosis is based on a constellation of physical findings, including six or more café-au-lait spots on the skin, freckling on the groin and underarms, development of tumor-like bumps on the skin (cutaneous neurofibromas) or larger areas along the nerves deeper in the body (plexiform neurofibromas), pigmented bumps on the iris, and skeletal manifestations like scoliosis or enlargement or deformity of bones.
Schwannomatosis
Less common types of NF are classified as schwannomatosis (SWN). What had been known as NF type 2 was reclassified in 2022 as NF2-related SWN (NF2-SWN), which is caused by genetic mutations on chromosome 22. Others are named for the gene change that causes them, such as SMARCB1-related SWN, LZTR1-related schwannomatosis, and 22q-related SWN. Some are also classified as SWN not otherwise specified or not elsewhere classified.
Together, the various types of SWN affect approximately one in 20,000 births; excluding NF2-SWN, that figure is around one in 70,000. While these conditions can emerge at any age, symptoms typically develop among teens and adults.
NF2-SWN arises from mutations, more often spontaneous than inherited, in a tumor-suppressing gene, leading to characteristic benign tumors on the cranial nerve that transports sound and balance input to the brain. These vestibular schwannomas are responsible for some of the most common symptoms: tinnitus, hearing loss, and impaired balance. NF2-SWN may also cause meningiomas, ependymomas, and juvenile cataracts.
Pain is commonly the presenting symptom. In one case series, chronic pain was noted to affect some 68% of patients, “and usually persisted despite aggressive surgical and medical management.”
Treatment Options
Large tumors or those pressing on the spine, brain, or other key structures may require surgery to reduce symptoms. Benign but bothersome cutaneous neurofibromas or skin schwannomas may be surgically removed as well.
Stereotactic radiosurgery or medications can help control pain.
Pharmaceutical options had been limited, though, until breakthroughs began in the 1990s, said Packer. First was understanding how NF causes tumors by allowing overactivity of the Ras-MAP kinase system. “Honestly, even as great as that was,” he said, “it would have been a very partial victory without knowing what to do with it. But there were a set of drugs that were being developed for melanoma that affected the Ras-MAP kinase pathway, which is also driven by abnormalities in that pathway.”
Drugs to inhibit the MAP kinase enzymes MEK1 and MEK2 block cell proliferation and induce apoptosis.
The MEK inhibitor selumetinib (Koselugo) became the first drug approved for NF in 2020, with an indication for reducing the size of inoperable plexiform neurofibromas. This was based on findings from a phase II trial in 50 children with NF1 and symptomatic inoperable plexiform neurofibromas, in which 68% had a confirmed partial response (at least 20% decrease in target lesion size) and tumor pain intensity improved by a clinically meaningful 2 points on a 10-point scale.
While other MEK inhibitors and further indications in NF for them are being developed, even having the one drug has been a turning point, Packer said.
“The MEK inhibitors were the first ones that were truly selective for the NF1 type of condition, and they have made a tremendous difference in the outcome of these patients,” he said. “The studies have been ongoing for about a decade, but obviously, the seal of approval of the FDA that [there is] a proven effective therapy convinced many patients who were not sure they wanted to be treated that they did want [treatment]. And now it’s also shifted the mindset of physicians taking care of patients with NF, that these things aren’t immutable, that we can make patients better, that we can make tumors shrink.”
And it shows that the efforts to build the machinery to study the condition and run clinical trials are paying off, said Bakker. “What you do see is you see a massive acceleration of research and development.”
Disclosures
Packer disclosed no relevant relationships with industry.
Bakker disclosed being president of the Children’s Tumor Foundation and also the chair of CTF Europe, which is a another nonprofit connected to the Children’s Tumor Foundation.
Please enable JavaScript to view the