
Dive Brief:
- The Food and Drug Administration on Friday said healthcare providers should not use Hologic’s Biozorb radiographic markers. Hologic recalled the devices in March due to reports of serious adverse events in patients who had them implanted in breast tissue.
- Hologic notified customers Thursday that 188 complaints were associated with adverse events and asked that all unused lots be returned to the company. Patients, however, do not need to have the devices explanted unless advised to by a healthcare provider, Hologic said.
- The FDA updated a February safety communication that warned of the potential for serious complications with the Biozorb markers. In May, the FDA classified Hologic’s recall as Class I, the most serious type, but the product was not removed from the market.
Dive Insight:
Hologic said it has received patient complaints that described pain, infection, rash, device migration, device erosion, fluid buildup and discomfort.
In some instances, additional medical treatment, including having the device removed from the body, was needed, the FDA said.
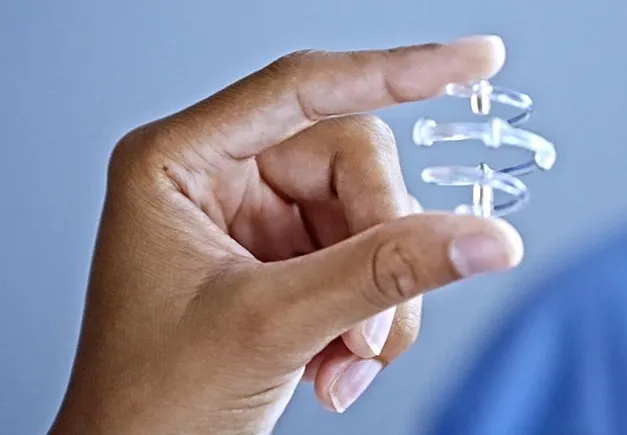
The implantable devices are used for radiographic marking of soft tissue sites, including breast tissue, and in situations where the soft tissue site needs to be marked for future medical procedures.
The markers consist of a plastic component designed to be dissolved in the patient’s body within a year or longer and a permanent titanium metal component.
In its latest letter to customers, Hologic said 91,531 of the markers have been sold since 2015. Of 399 complaints about the devices, 188 were associated with adverse events as of Oct. 16, 2024.
The earlier recall identified 71 reported injuries and no reports of death.
Hologic said it is working closely with the FDA on the voluntary product removal.